

Abstract
Our group developed a 161533 trispecific killer engager (TriKE) molecule to target acute myeloid leukemia (AML) cells using Natural Killer (NK) cells. This molecule contains an anti-CD16 camelid nanobody to activate NK cells, an anti-CD33 single chain variable fragment (scFv) to engage cancer targets, and an IL-15 molecule that drives NK cell priming, expansion and survival. Using an earlier version of this molecule, we have shown that the CD33 TriKE is effective at activating NK cells against AML targets in vitro and in vivo. This preclinical data has lead to the establishment of a clinical trial in refractory AML patients at the University of Minnesota, set to open Q3 2018. While these previous studies have validated the use of TriKEs as an effective strategy of harnessing NK cells in cancer immunotherapy, CD33 has limitations as a target antigen.
The high mortality and poor five-year survival rates (26%) for AML patients can be attributed to chemotherapy resistance and disease relapse. A majority of chemotherapy resistant leukemia stem cells (LSCs), that are hypothesized to facilitate relapse, do not express CD33. In addition, all hematopoietic stem cells and normal myeloid cells express CD33, thus targeting this antigen can lead to severe defects in hematopoiesis and on-target/off-tumor toxicity. To address these limitations, we developed a TriKE that targets CLEC12A or C-type lectin-like molecule 1 (CLL-1). CLEC12A is highly expressed on AML cells and over 70% of CD33 negative cells express CLEC12A. It has been attributed as a stem cell marker in AML, being selectively overexpressed in LSCs. CLEC12A is expressed by CD34+/CD38- LSCs but not normal CD34+/CD38- hematopoietic stem cells in regenerating bone marrow, thus minimizing off-target effects.
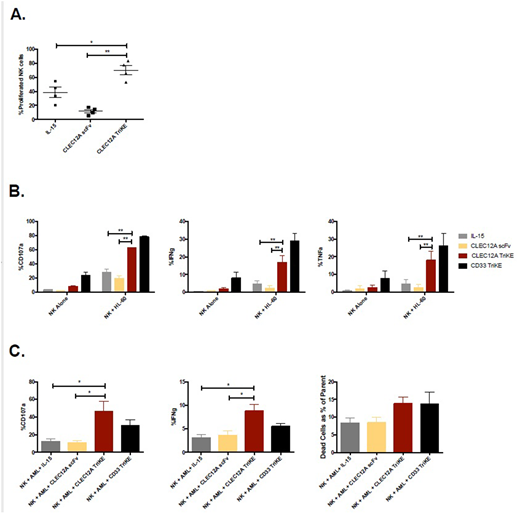
The CLEC12A TriKE was developed in a mammalian cell system to ensure that appropriate post-translational modifications are present. We confirmed that the TriKE binds specifically to HL-60 and THP-1 target cells that express CLEC12A compared to Raji cells that do not express CLEC12A. Treatment of peripheral blood mononuclear cells (PBMCs) with the CLEC12A TriKE drives a significant increase in NK cell specific proliferation over 7 days as measured by CellTrace dilution compared to treatment with a CLEC12A scFv or IL-15 alone (69.7 ± 6.7% vs 11.9 ± 2.5% vs 38.4 ± 7.3%) (Figure 1A). To measure NK cell killing, we conducted an IncuCyte Zoom assay. Here, HL-60 target cells were labeled with a caspase 3/7 reagent where a color change indicates target cell death. The CLEC12A TriKE was able to induce more target cell killing than CLEC12A scFv or IL-15 as measured by number of live target cells at the end of the 48 hour assay (53.9 ± 1.9% vs 103.3 ± 3.4% vs 71.1 ± 1.4%). The CLEC12A TriKE induces an increase in NK cell degranulation, measured by CD107a expression against HL-60 AML tumor targets in a 4 hour functional assay compared to treatment with CLEC12A scFv or IL-15 alone (62.3 ± 1.1% vs 19.4 ± 3.8% vs 27.5 ± 4.9%). In this assay, there is also an increase in cytokine production, measured by IFNg and TNFa respectively (16.7 ± 4.2% vs 2.3 ± 1.5% vs 4.7 ± 1.9% and 18.0 ± 5.1% vs 2.5 ± 1.7% vs 4.6 ± 2.5%) (Figure 1B). We observe a similar enhanced functional response with THP-1 AML tumor targets. In these functional assays, treatment with the CLEC12A TriKE produced less background activation compared to the CD33 TriKE, indicating less off-target effects on PBMCs. To confirm the clinical relevance of this molecule, we tested the efficacy of the CLEC12A TriKE against primary AML targets. AML blasts were identified as SSC low, CD45 intermediate and CD34 high cells. Out of the 9 AML samples tested, 7 expressed high levels of CD33 (70.4 ± 6.3%) and CLEC12A (78.1 ± 5.2%). In functional assays with these samples, the CLEC12A TriKE was able to induce greater CD107a and IFNg expression, and enhanced killing of tumor targets as measured by a live/dead stain compared to CLEC12A scFv or IL-15 (Figure 1C). In these assays, the efficacy of the CLEC12A TriKE was comparable to the CD33 TriKE. Our data demonstrates that the CLEC12A TriKE drives NK cell specific proliferation, degranulation, cytokine secretion, and killing of tumor targets in vitro. Apart from AML, CLEC12A is expressed on cancer cells and LSCs in patients with myelodysplastic syndromes (MDS). These findings highlight the clinical potential of the CLEC12A TriKE individually or in combination with the CD33 TriKE for the treatment of MDS and AML.
Vallera:GT Biopharma: Consultancy, Research Funding. Felices:GT Biopharma: Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal